La fibrosi cistica, o mucoviscidosi, è la malattia genetica autosomica recessiva con prognosi infausta più frequente nelle popolazioni caucasiche in Europa, Nord America e Australia e affligge approssimativamente 1 neonato ogni 3500 nati vivi.
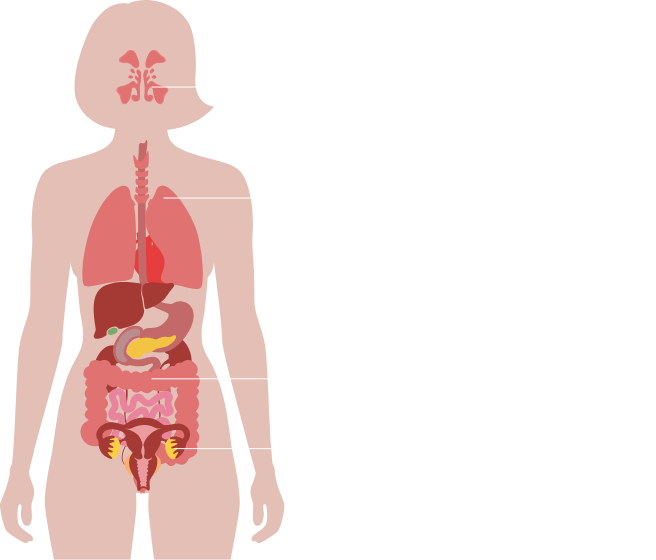
E’ caratterizzata da un eccesso di secrezioni a livello di tutte le ghiandole esocrine, i cui effetti più gravi e frequenti sono a carico dell’apparato respiratorio e gastrointestinale.
La fibrosi cistica è la principale causa di malattia respiratoria cronica nel bambino ed è responsabile della maggior parte delle insufficienze pancreatiche nell’infanzia. È inoltre responsabile di molti casi di disidratazione con perdita di sali, poliposi nasale, pansinusite, prolasso rettale, pancreatite, colelitiasi, epatopatia cronica e diabete.
La malattia è causata da mutazioni nel gene che codifica per la proteina CFTR (Cystic Fibrosis Transmembrane Regulator), una proteina transmembrana che regola il trasporto del cloro, del sodio e del bicarbonato attraverso le membrane epiteliali.
L’assenza, una ridotta quantità o il malfunzionamento della proteina CFTR determina una carenza di cloro e acqua nelle secrezioni che pertanto diventano eccessivamente dense, disidratate e viscose.
Nelle vie aeree la formazione e il ristagno eccessivo di muco favorisce l’insorgenza di infezioni respiratorie ricorrenti, anche da parte di batteri opportunisti, e uno stato infiammatorio ingravescente che con il tempo porta un pericoloso deterioramento dell’albero bronchiale e del tessuto polmonare e quindi alla perdita della funzionalità respiratoria.
Bronchiectasia, ostruzione delle piccole vie aeree e insufficienza respiratoria progressiva rappresentano le principali cause di morte nei pazienti affetti da fibrosi cistica.
Oltre alla patologia polmonare cronica, la fibrosi cistica si associa a insufficienza pancreatica esocrina, disturbi epatobiliari, interessamento delle vie aeree superiori (naso e seni paranasali) e dell’apparato riproduttivo e livelli particolarmente elevati di elettroliti nel sudore.
Nel 1950 i bambini affetti da fibrosi cistica morivano pochi mesi dopo la nascita per ileo da meconio o malnutrizione dovuta a malassorbimento pancreatico e per molti anni la fibrosi cistica è rimasta una malattia tipicamente pediatrica.
Da allora, la comprensione dei meccanismi patogenetici e la definizione del percorso diagnostico-terapeutico ha contribuito a migliorare il quadro clinico, la qualità di vita e l’aspettativa di vita dei pazienti affetti grazie a
– diagnosi precoce della malattia,
– trattamenti che consentono di prevenire l’ostruzione delle vie aeree e di controllare le infezioni respiratorie, di correggere l’insufficienza pancreatica e la malnutrizione
– terapie innovative recenti che modulano la conduttanza transmembrana del CFTR.