La diagnosi di fibrosi cistica si basa su tre elementi fondamentali:
l’esistenza di un quadro clinico compatibile con la malattia, rilevato attraverso la storia clinica del paziente e l’esame obiettivo o lo screening neonatale positivo
la disfunzione del canale ionico di trasporto del cloruro e del bicarbonato Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), misurata attraverso il test del sudore
l’identificazione di mutazioni genetiche del CFTR attraverso il test genetico.
Tabella. Tecniche di terapia drenante [tratta da Borgo]
Valutazione clinica
In presenza di almeno uno dei seguenti elementi rilevati dalla storia clinica e dell’esame obiettivo è opportuno eseguire un test del sudore in bambini e giovani o un test genetico negli adulti:
Storia familiare
Atresia intestinale congenita
Ileo da meconio
Sintomi e segni da sindrome dell’ostruzione intestinale distale
Ritardo di crescita in neonati e bambini piccoli
Malnutrizione in bambini più grandi e adulti
Malattia polmonare ricorrente e cronica
– infezioni ricorrenti delle basse vie respiratori
– dati clinici e/o radiologici di patologie polmonari, in particolare bronchiectasie
– modifiche radiografiche persistenti del torace-addome
– tosse cronica umida o produttiva
Sinusite cronica
Azoospermia ostruttiva nei giovani e negli adulti
Pancreatite acuta o cronica
Malassorbimento
Prolasso rettale nei bambini
Sindrome di pseudo-Bartter
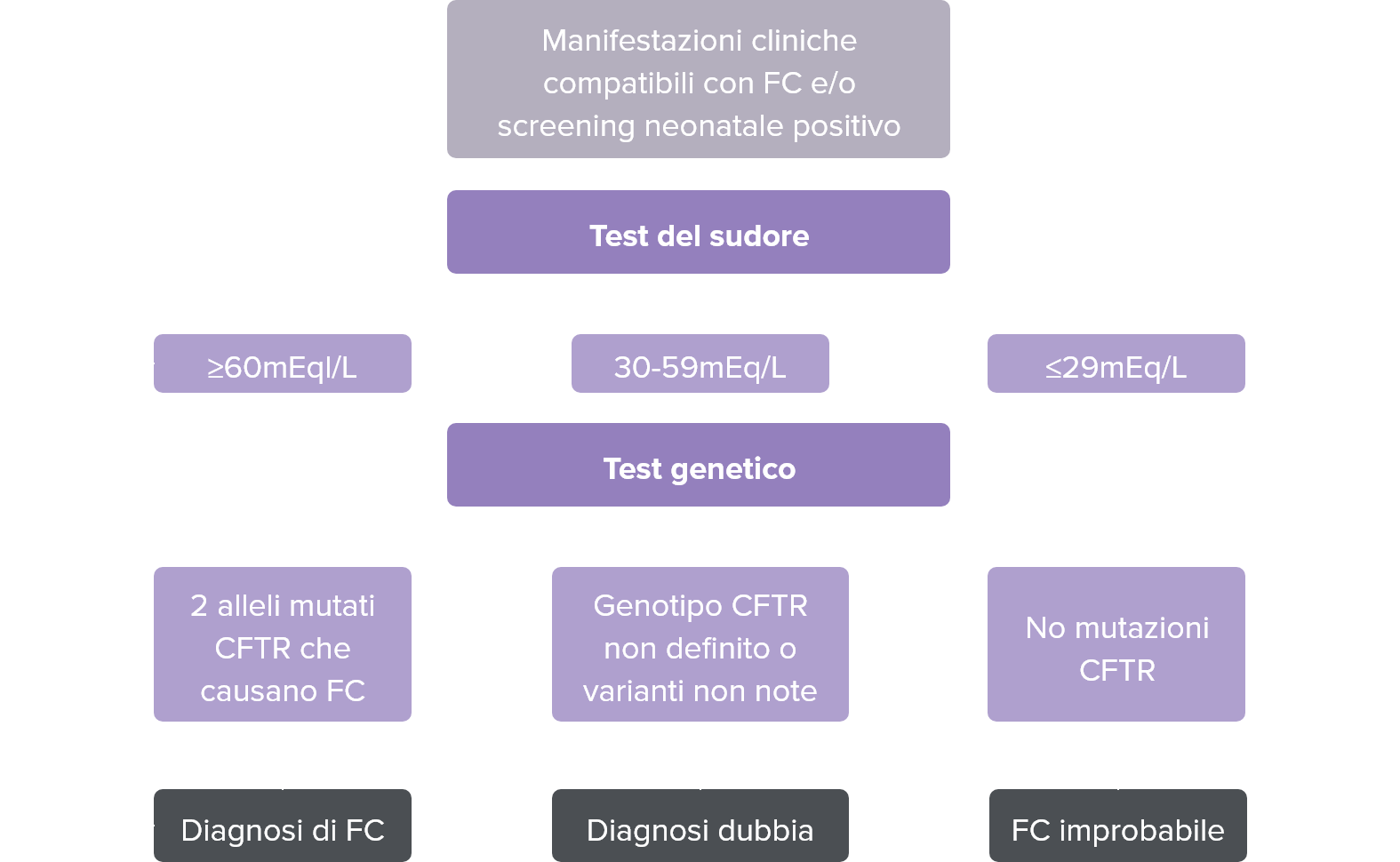
Test del sudore
Il test del sudore rappresenta il gold standard per la diagnosi di fibrosi cistica. E’ l’unico test in grado di misurare in modo riproducibile il difetto di funzionamento della proteina di membrana CFTR, che determina un’elevata concentrazione di ione cloro nel sudore.
Secondo i dati del Registro Italiano nel 2018 il 92,6% dei pazienti con diagnosi di fibrosi cistica ha eseguito almeno un test del sudore mediante dosaggio del cloro. Il 41,5% dei pazienti è stato sottoposto anche al test per il dosaggio del NaCl.
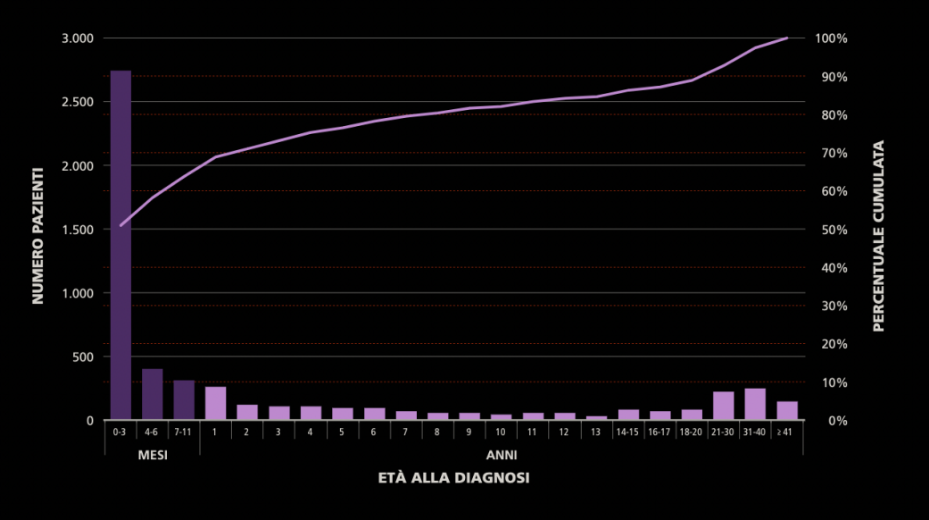
La maggior parte dei pazienti riceve la diagnosi entro il secondo anno di vita (70,9%), senza distinzioni significative tra maschi e femmine. La quota di diagnosi effettuate in età adulta non è tuttavia trascurabile e costituisce il 18,3% della popolazione totale nel 2018. I soggetti con mutazioni rare o manifestazioni atipiche (es. sintomi respiratori lievi, sterilità maschile) possono raggiungere l’età adulta senza diagnosi. Questo dato sottolinea la necessità di una maggiore sensibilità da parte della comunità medica a porre il sospetto di fibrosi cistica anche in età adulta.
Figura. Età alla diagnosi dei pazienti con fibrosi cistica presenti nel Registro Italiano Fibrosi Cistica. Anno 2018
Quando fare il test del sudore
Le indicazioni per eseguire il test del sudore includono:
fenotipo suggestivo di FC
storia familiare di FC
screening neonatale per FC positivo
fenotipo suggestivo di patologia correlata ad FC
Il test deve essere eseguito in strutture specializzate.
Efficacia diagnostica del test del sudore
Il test del sudore trova ampia applicazione e assume elevata specificità diagnostica quando è utilizzato per la diagnosi di fibrosi cistica in presenza di almeno una manifestazione clinica tipica e un test del sudore con valori superiori a 60 mEq/L di cloro sudorale.
Il test del sudore non assume significato discriminatorio nella diagnosi delle CFTR-RD (CFTR-related disease) o CF-SPID (screening positivo, diagnosi non conclusiva), nelle quali le concentrazioni di cloruro si attestano in genere nella fascia dubbia (nella quale può comunque essere di ausilio diagnostico) o addirittura negativa.
La diagnosi di fibrosi cistica può rimanere incerta in quei pazienti con caratteristiche cliniche suggestive della patologia, test del sudore intermedio o negativo e 1 o 2 mutazioni non identificate. In questi casi saranno utili per la diagnosi ulteriori studi della funzione della proteina (misura dei potenziali nasali o delle correnti intestinali).
In base agli esiti del test i soggetti con valori elevati vengono inviati ai centri specialistici per ulteriori accertamenti e l’inizio del trattamento.
Analisi genetica per la fibrosi cistica
Ad oggi sono disponibili diverse metodologie di analisi basate su tecniche molecolari che permettono di individuare varianti del gene CFTR. Queste tecniche utilizzano DNA (o in alcuni casi RNA) estratto da diverse tipologie di campione biologico (es: prelievo di sangue, tampone buccale, spot di sangue, brushing nasale).
L’analisi molecolare è offerta da laboratori che operano in associazione con servizi di Genetica Clinica, Centri di Riferimento per la fibrosi cistica e Servizi di supporto.
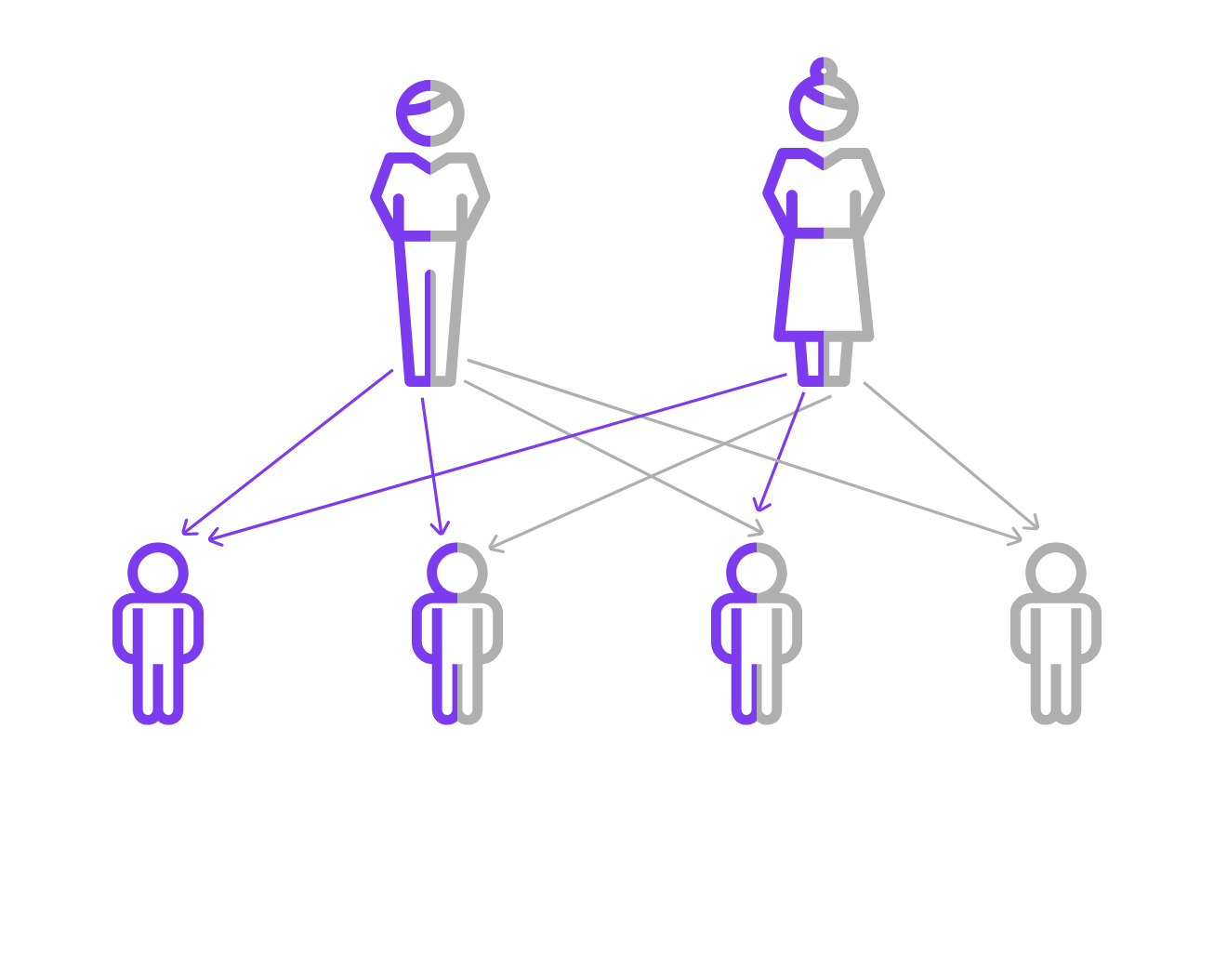
In quanto malattia autosomica recessiva, la fibrosi cistica si manifesta nei soggetti che ereditano due copie entrambe mutate del gene CFTR. In caso di una sola copia mutata del gene la persona interessata è un portatore sano, privo dei sintomi caratteristici della malattia.
Per la diagnosi di fibrosi cistica è auspicabile dimostrare la presenza di due alleli mutati, motivo per cui l’analisi molecolare del gene CFTR è sempre indicata.
Nel caso in cui il test del sudore risulti positivo, l’analisi molecolare ha un ruolo di conferma.
Quando non è possibile eseguire l’esame del sudore o quando l’esito di questo test risulta negativo o di dubbia interpretazione, in caso di sospetto clinico l’analisi molecolare del gene CFTR assume un ruolo dirimente.
In caso di disfunzioni del CFTR che non rispettano i criteri diagnostici della fibrosi cistica o in caso di positività allo screening neonatale con diagnosi non conclusiva, è opportuno considerare l’eventualità di Sindrome Metabolica CFTR-Correlata (CRMS) o di patologie correlate al CFTR (CFTR-RD).
L’indagine genetica rende anche possibile l’eventuale test del portatore e la diagnosi prenatale all’interno della famiglia del paziente. Determinare il genotipo del paziente serve inoltre a identificare i soggetti che possono beneficiare di interventi terapeutici mirati essendo portatori di quelle varianti patogenetiche per le quali sono disponibili farmaci specifici.
Indagini mutazionali per la fibrosi cistica
test di I livello: indagano le varianti patogenetiche più frequenti del gene CFTR attraverso tecniche semplici e rapide (es: Reverse Dot Blot, Amplification Refractory Mutation System)
test di II livello: indagano la sequenza di tutti gli esoni, delle zone introniche adiacenti gli esoni, delle regioni del promotore, del 3’ non tradotto e delle regioni pienamente introniche, sedi di varianti patogenetiche di splicing conosciute del gene CFTR. Permettono inoltre di individuare un maggior numero di varianti, anche rare e non note, grazie a tecniche quali il metodo Sanger e il Next Generation Sequencing
test di III livello: indagano grossi riarrangiamenti del gene CFTR (macrodelezionzi/macroduplicazioni), compresi quelli non evidenziabili dai test precedenti, servendosi di tecniche specifiche (es: Multiplex ligation dependent probe amplification, Real Time PCR)
test di IV livello: indagano varianti non conosciute che interferiscono con la maturazione dell’mRNA del gene CFTR (es: Reverse transcriptase PCR)
Screening neonatale per la fibrosi cistica
Lo screening neonatale ha l’obiettivo di individuare entro i due mesi di vita i neonati con aumentata probabilità di malattia per sottoporli il prima possibile all’iter diagnostico.
Alla nascita viene eseguito un piccolo prelievo ematico per il dosaggio su cartoncino di Guthrie del tripsinogeno immunoreattiovo IRT, un enzima pancreatico che risulta elevato nei neonati affetti.
In caso si rilevazione di valori oltre la soglia di normalità, il test viene ripetuto una seconda volta.
I neonati risultati positivi anche al secondo dosaggio vengono indirizzati presso i Centri di Riferimento Regionali per la fibrosi cistica per gli accertamenti opportuni per la diagnosi definitiva:
test del sudore,
valutazione clinica
eventuale analisi genetica
Secondo il Registro italiano Fibrosi Cistica, i pazienti diagnosticati entro i primi 24 mesi di vita grazie allo screening neonatale sono in continuo aumento, grazie all’implementazione di appositi programmi di screening in quasi tutte le regioni italiane, che oggi coprono il 96% della popolazione neonatale italiana.
Lo screening neonatale per la fibrosi cistica è diventato obbligatorio alla nascita grazie alla legge n. 104 del 5 febbraio 1992.
Diagnosi prenatale
La diagnosi prenatale per la fibrosi cistica consiste in un test molecolare condotto su materiale fetale prelevato attraverso amniocentesi o villocentesi.
Il test viene eseguito quando il feto è ritenuto a rischio di fibrosi cistica, in primo luogo nel caso in cui entrambi i genitori siano portatori noti di una variante patogenetica del gene CFTR (genitori eterozigoti) e quando uno dei genitori è malato.
Una coppia di adulti eterozigoti per una variante del gene CFTR ha una probabilità del 25% (1/4) di concepire un figlio affetto da fibrosi cistica.
Le coppie di portatori possono essere individuate attraverso l’analisi genetica di familiari affetti, familiari di portatori o anche di individui della popolazione generale.
Nel caso in cui sia note le varianti patogenetiche di entrambi i soggetti portatori, nel feto si procede innanzitutto con la ricerca delle mutazioni patogenetiche dei genitori (analisi diretta).
Test del portatore
Il test del portatore prevede l’analisi molecolare mirata alla ricerca di eventuali mutazioni del gene CFTR. Viene eseguito su un singolo individuo previo consenso informato.
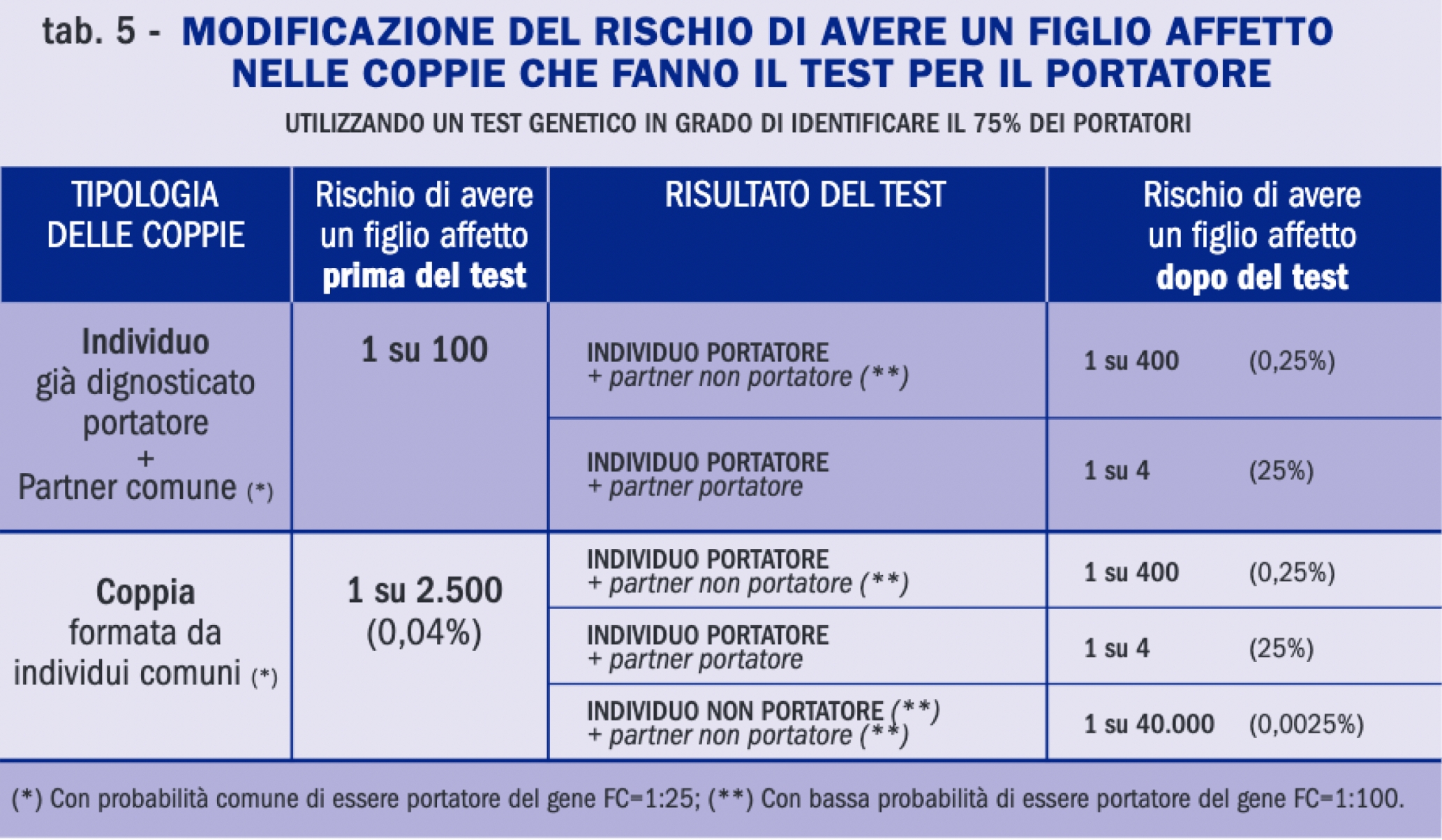
I test genetici oggi disponibili permettono di individuare con sensibilità elevata, ma assoluta, i portatori di mutazioni del gene CFTR, che si presentano completamente asintomatici.In caso di positività il soggetto viene indirizzato verso una successiva consulenza genetica specialistica; se invece il test ha esito negativo, il referto riporterà l’indicazione di rischio residuo (la probabilità che lo screening non abbia identificato una mutazione presente).
Oggi l’offerta dello screening a cascata sui familiari del soggetto malato permette di individuare con efficienza i portatori sani di fibrosi cistica. Al momento, in Italia, la situazione normativa è fortemente differenziata nelle diverse regioni; nella maggior parte dei casi il test del portatore è a carico del Sistema Sanitario Nazionale e accessibile in caso di familiarità stretta con un malato.
In assenza di familiarità positiva per la fibrosi cistica per il test del portatore è indicata l’analisi genetica di I livello.
Tabella. RIschio di avere un figlio affetto nelle coppie che fanno il test del portatore
Bibliografia
Analisi genetica in Fibrosi Cistica, Società Italiana per lo studio della fibrosi cistica (SIFC), Consensus 2019
Cartabellotta A, Furnari M, Linee guida per la diagnosi e la terapia della fibrosi cistica. Evidence 2018; Volume 10 (Issue 1)e1000176
Registro Italiano Fibrosi Cistica (RIFC), Rapporto 2017-2018. Epidemiol Prev 2019; 45 (3) Suppl 1:1-37. doi: 10.19191/EP21.3.S1.050
Farrell P M et al, Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. The Journal of Pediatrics, Volume 181S, February 2017
Kessels S J M et al, Prenatal genetic testing for cystic fibrosis: a systematic review of clinical effectiveness and an ethics review. Genetics in Medicine volume 22, pages258–267 (2020
Raccomandazioni sul test del portatore di mutazioni del gene CTFR, Società Italiana per lo studio della fibrosi cistica (SIFC)