Al momento non esiste una terapia che consente la guarigione dalla fibrosi cistica, tuttavia l’impostazione di un programma terapeutico personalizzato e globale, in grado di affrontare le diverse sfaccettature cliniche della malattia, unitamente alle più recenti terapie innovative, negli anni ha prolungato significativamente la sopravvivenza dei pazienti FC.
Infatti, la fibrosi cistica, se inquadrata correttamente e affrontata precocemente, non è più una malattia unicamente pediatrica, ma interessa anche la medicina dell’adulto.
In base alla legge nazionale n. 548/1993 i malati FC possono rivolgersi a centri specializzati per la diagnosi e la cura della fibrosi cistica presenti in ogni regione d’Italia.
Ogni centro provvede a un approccio multidisciplinare dei pazienti con team composti da medici, infermieri, fisioterapisti, nutrizionisti, professionisti della salute mentale, farmacisti e assistenti sociali, psicologi e consulenti per il trattamento di particolari complicanze.
Sul sito della Società Italiana Fibrosi Cistica (SIFC) è possibile consultare la lista completa dei Centri FC italiani.
Obiettivo principale della terapia è quello di favorire la crescita fisica e psicosociale del paziente con fibrosi cistica, rallentando il progredire della malattia e prevenendo l’insorgenza delle complicanze d’organo.
Trattamento sintomatico complessivo della fibrosi cistica:[tratto da Dtsch Aeztebl Int 2017]
• terapia digestivo-nutrizionale
– terapia enzimatica sostitutiva
– nutrizione ipercalorica e ad alto contenuto di grassi
– supplementazione vitaminica
– acido ursodesossicolico
– prevenzione/trattamento occlusione intestinale
• terapia respiratoria
– antibiotici
– mucolitici
– antinfiammatori
– lavaggi nasali
– fisioterapia respiratoria
• modulatori della proteina CFTR
La somministrazione di enzimi pancreatici e l’impostazione di un piano nutrizionale adeguato rappresentano uno dei cardini della terapia dei pazienti con fibrosi cistica. La riduzione o l’assenza di enzimi pancreatici endogeni, dovuti all’insufficienza pancreatica presente in circa l’85% dei pazienti, rende impossibile l’assorbimento dei nutrienti e quindi espone i pazienti a un elevato rischio di malnutrizione grave, con conseguente rallentamento della crescita e rischio di complicanze.
La mancanza di enzimi pancreatici è particolarmente dannosa per la digestione dei grassi. Senza l’azione degli enzimi, i grassi introdotti non vengono digeriti ma persi con le feci, che diventano frequenti, unte, poco solide e abbondanti.
La terapia enzimatica sostitutiva deve essere somministrata con l’assunzione dei pasti e degli spuntini a un dosaggio personalizzato compreso tra 2000-4000 U lipasi/Kg di peso corporeo/pasto (8000-10.000 U lipasi/kg di peso corporeo/die) che dovrebbe essere calcolato in base al contenuto di grassi del pasto e delle caratteristiche del paziente (peso, altezza ed età) e viene suggerito dai centri di riferimento.
Un dosaggio troppo ridotto di enzimi può non essere in grado di correggere il difetto e quindi non consente di migliorare la perdita di grassi con le feci che rimangono oleose. Al contrario, l’utilizzo di dosaggi troppo elevati di enzimi pancreatici può favorire l’insorgenza di infiammazione cronica del colon, la cui parete si inspessisce e diventa meno elastica rendendo più difficile la progressione delle feci nell’intestino.
Nei pazienti che necessitano di dosaggi elevati di enzimi pancreatici, l’efficacia del trattamento può essere migliorata dalla soppressione della secrezione acida indotta da anti-H2 o inibitori della pompa protonica.
Il paziente affetto da fibrosi cistica, a maggior ragione in presenza di insufficienza pancreatica, presenta un elevato rischio di malnutrizione per malassorbimento. Per questo motivo, è fondamentale impostare un piano nutrizionale personalizzato caratterizzato da un’alimentazione ipercalorica e ricca di grassi per garantire la crescita e lo sviluppo dei bambini, un miglior funzionamento del sistema immunitario, una migliore funzione respiratoria e in genere un miglior andamento della malattia con riduzione delle complicanze.
La terapia alimentare del paziente affetto da fibrosi cistica comprende:
• nutrizione ad alto apporto calorico superiore del 20-50% rispetto all’introito raccomandato per soggetti della stessa età e sesso e di grassi:
– proteine 20% delle calorie totali
– lipidi 40% delle calorie totali
– glucidi 40-50% delle calorie totali, di cui 10% zuccheri semplici.
• supplementazione multivitaminica ADEK in quantità doppia rispetto ai fabbisogni giornalieri raccomandati
• supplementazione di sali minerali, compresi fluoro e zinco, soprattutto nei bambini più piccoli e nei periodi di stress termico
Per quanto riguarda l’alimentazione dei neonati, l’allattamento al seno non solo è consentito ma incentivato con l’accortezza della supplementazione di sali minerali per scongiurare la disidratazione. Nei neonati allattati artificialmente è necessario optare per formule con maggiore apporto calorico e un’elevata concentrazione di sali minerali, di vitamine liposolubili e di grassi digeribili. Per aumentare l’apporto calorico possono essere somministrati polimeri di glucosio e acidi grassi a catena media.
L’integrazione enterale mediante gastrostomia o digiunostomia può migliorare la crescita e il mantenimento della funzione respiratoria nei pazienti che non riescono a mantenere uno stato nutrizionale adeguato.
Gli stimolatori dell’appetito possono essere di aiuto.
Nei neonati l’occlusione intestinale può essere risolta con un clisma con mezzo di contrasto radiopaco iperosmolare o isomolare, o in caso di necessità attraverso l’enterostomia chirurgica.
Nei bambini e negli adulti l’occlusione intestinale parziale può essere trattata con un clisma con mezzo di contrasto radiopaco iperosmolare o isomolare oppure con acetilcisteina oppure con soluzioni di lavaggio intestinale.
A titolo preventivo e per favorire il transito delle feci può essere utile la somministrazione di emollienti fecali.
Infine, la somministrazione di acido ursodessicolico può migliorare il quadro epatico in alcuni pazienti con malattia epatica correlata a fibrosi cistica. Il trattamento deve essere prescritto dopo accurata valutazione dei parametri ematochimici ed ecografici.
Il ristagno di muco e le secrezioni dense e viscose nelle vie respiratorie espongono i pazienti affetti da fibrosi cistica a infezioni bronco-polmonari ricorrenti.
I germi più comunemente isolati sono:
La terapia antibiotica, definita in base all’antibiogramma, rappresenta pertanto una strategia fondamentale nel trattamento delle infezioni batteriche acute sia delle esacerbazioni di infezioni croniche del paziente con fibrosi cistica.
Il dosaggio utilizzato è generalmente più elevato nei pazienti con fibrosi cistica rispetto alla popolazione generale.
Le principali vie di somministrazione antibiotica sono per via inalatoria ed endovenosa, e orale per le infezioni acute.
La somministrazione antibiotica per via orale è impiegata principalmente nelle infezioni acute e nelle riacutizzazioni più lievi. Purtroppo solo pochi antibiotici attivi contro lo P. aeruginosa e altri microrganismi tipici della FC presentano un buon assorbimento intestinale.
La via inalatoria è principalmente utilizzata nel trattamento delle infezioni croniche e offre il vantaggio di raggiungere più efficacemente e in modo mirato le vie aeree con minor rischio di effetti collaterali a livello di altri apparati, quali reni e fegato.
La somministrazione per via endovenosa è la modalità più efficace, perché consente di ottenere le più elevate concentrazioni di antibiotici nei polmoni, ed è indispensabile soprattutto in caso di lesioni polmonari importanti e nelle esacerbazioni in corso di infezione respiratoria cronica.
In alcuni centri la cura della malattia respiratoria “cronica” prevede cicli di terapia antibiotica per via endovenosa, programmati a cadenza trimestrale o quadrimestrale (indipendentemente dai sintomi), eventualmente associata ad aerosol antibiotico continuativo a mesi alterni.
La terapia inalatoria è un appuntamento fisso e fondamentale per il paziente affetto da fibrosi cistica e prevede l’impiego di diverse categorie di farmaci:
L’aerosolterapia è sicuramente una terapia impegnativa che richiede una forte motivazione da parte del paziente. Si calcola che il paziente con fibrosi cistica dedichi fino a 4 ore al giorno per la fisioterapia respiratoria e le terapie inalatorie. Proprio per il suo impatto significativo sulla vita dei pazienti, nonostante i benefici dimostrati, è necessario rinforzare la motivazione e favorire l’aderenza al trattamento.
Nei bambini piccoli è necessaria la collaborazione di un familiare, nei più grandi è fondamentale l’istruzione all’autogestione degli strumenti e della modalità di esecuzione.
Alcune regole generali per migliorare l’efficacia dell’aerosolterapia:
È stato dimostrato che la terapia quotidiana con mucolitici, in particolare DNase per via inalatoria o una soluzione salina ipertonica al 7%, riduce la velocità di decadimento della funzionalità respiratoria e diminuisce la frequenza delle esacerbazioni.
La fibrosi cistica è caratterizzata da una situazione clinica in cui si combinano infezione e infiammazione delle vie aeree, che periodicamente riacutizzano. La malattia FC esprime già nei primi due anni di vita una precoce infiammazione-infezione con formazione di bronchiectasie. La risposta infiammatoria a livello polmonare, con grande accumulo di neutrofili nelle vie aeree, determina il rilascio di proteasi e citochine (IL-8, IL-6, Tumor Necrosis Factors-alfa TNF) che danneggiano le pareti bronchiali incrementando la migrazione di neutrofili in un circolo vizioso. Numerosi studi stanno valutando l’efficacia del trattamento con antinfiammatori.
I corticosteroidi per via orale sono indicati nei lattanti con forme protratte di bronchiolite e nei pazienti con broncospasmo refrattario, aspergillosi broncopolmonare allergica e complicanze infiammatorie. L’impiego a lungo termine della terapia corticosteroidea a giorni alterni può rallentare il declino della funzionalità respiratoria, tuttavia non è raccomandata come trattamento di routine a causa delle possibili complicanze notoriamente legate agli steroidi (diabete, ritardo nella crescita e osteoporosi).
Anche l’uso cronico di antinfiammatori non steroidei (FANS) è stato oggetto di numerosi studi, che hanno dimostrato come la somministrazione prolungata soprattutto nei bambini tra i 5 e i 13 anni diminuisce la migrazione a livello polmonare dei neutrofili e rallenta il declino della funzionalità polmonare. Il rischio di eventi avversi gastrointestinali e renali correlati all’uso dell’antinfiammatorio richiede particolare cautela nell’utilizzo prolungato e stimola la ricerca di nuovi principi attivi (es. inibitore dell’elastasi) in grado di ridurre lo stato infiammatorio dei pazienti con fibrosi cistica.
La fisioterapia respiratoria si propone di mantenere le vie aeree libere dalle secrezioni migliorando la capacità di respiro, l’assorbimento delle terapie inalatorie e riducendo il ristagno di muco che può favorire l’insorgenza di infezioni.
Esistono diverse tecniche di fisioterapia respiratoria (vedi tabella).
È importante che il trattamento venga discusso e personalizzato in base alle esigenze e caratteristiche del singolo paziente, tenendo conto della capacità e possibilità di collaborazione, del danno polmonare, della presenza di complicanze, e dello stile di vita.
Per poter essere efficace è fondamentale che la fisioterapia respiratoria venga eseguita con regolarità quotidianamente, combinando anche più pratiche nella giornata.
Drenaggio posturale: si sfruttano i cambiamenti della posizione del corpo e la forza di gravità per favorire il drenaggio delle secrezioni da particolari zone del polmone, aiutandosi con la tosse.
Percussioni toraciche: sul torace vengono eseguite leggere percussioni con la mano a coppa per per aiutare le secrezioni a “sciogliersi”; questa tecnica è solitamente utilizzata insieme al drenaggio posturale nei bambini piccoli, ma è efficace a qualunque età.
Drenaggio autogeno: utilizza il controllo della respirazione per migliorare il flusso di aria espirata in modo che trascini con se il muco in eccesso.
Tecnica di espirazione forzata (FET): sfrutta la compressione dinamica del torace per far “risalire” il muco attraverso ripetuti atti di espirazione forzata a glottide semichiusa.
Ciclo attivo di tecniche respiratorie: si basa su 4 fasi di respirazione: respiro rilassato, respiro profondo, sbuffamento, tosse.
Tosse guidata: si basa sull’utilizzo di colpi di tosse volontari eseguiti in sequenza per drenare porzioni sempre più distali e profonde delle vie aeree. In questo modo si ottiene una sorta di “spremitura progressiva” delle vie aeree.
PEP mask: la maschera crea resistenza al flusso aereo alla bocca; espirando lentamente contro questa resistenza le vie respiratorie rimangono “aperte” facilitando l’eliminazione delle secrezioni. È ben tollerata e applicabile a qualsiasi età.
Flutter: si espira lentamente contro una resistenza oscillante determinata da una sfera che alternativamente apre o chiude il sistema: si ottengono così variazioni di pressione all’interno dei bronchi utili alla rimozione dei secreti.
Lavaggio nasale: è una tecnica molto importante per favorire la pulizia delle cavità nasali. La pratica più comunemente adottata, cui si dovrebbe abituare il malato fin dai primi mesi di vita, è quella dell’irrigazione nasale.
Attività fisica: anche l’attività fisica aerobica può essere considerata una modalità di fisioterapia di indiscussa efficacia, purché eseguita con regolarità e associata ad altre tecniche quando necessario. L’esercizio fisico favorisce lo sviluppo armonico del paziente, rinforza la muscolatura toraco-addominale, favorisce la rimozione delle secrezioni e contribuisce a una buona funzione respiratoria. E’ pertanto importante stimolare i pazienti fin da piccoli al movimento e al gioco attivo.
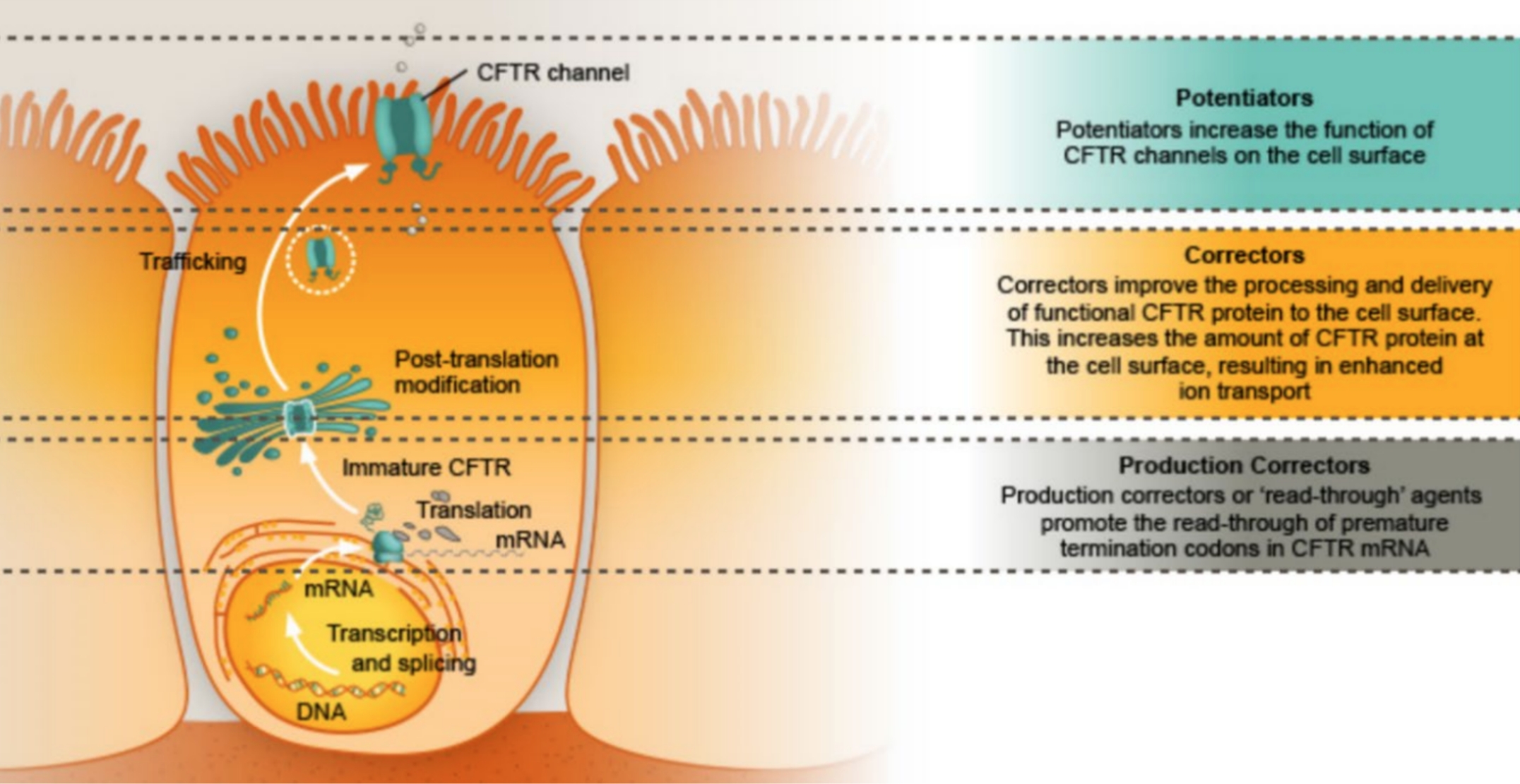
Accanto alla terapia sintomatica della fibrosi cistica, la ricerca da diversi anni rivolge i suoi sforzi nell’identificazione di soluzioni che possano correggere il difetto genetico all’origine della malattia (terapia genica) o recuperare la funzionalità della proteina CFTR mutata.
Mentre i risultati della terapia genica al momento non consentono di prenderla in considerazione come strategia di cura, si sono aggiunti all’armamentario del clinico i cosiddetti modulatori della proteina CFTR.
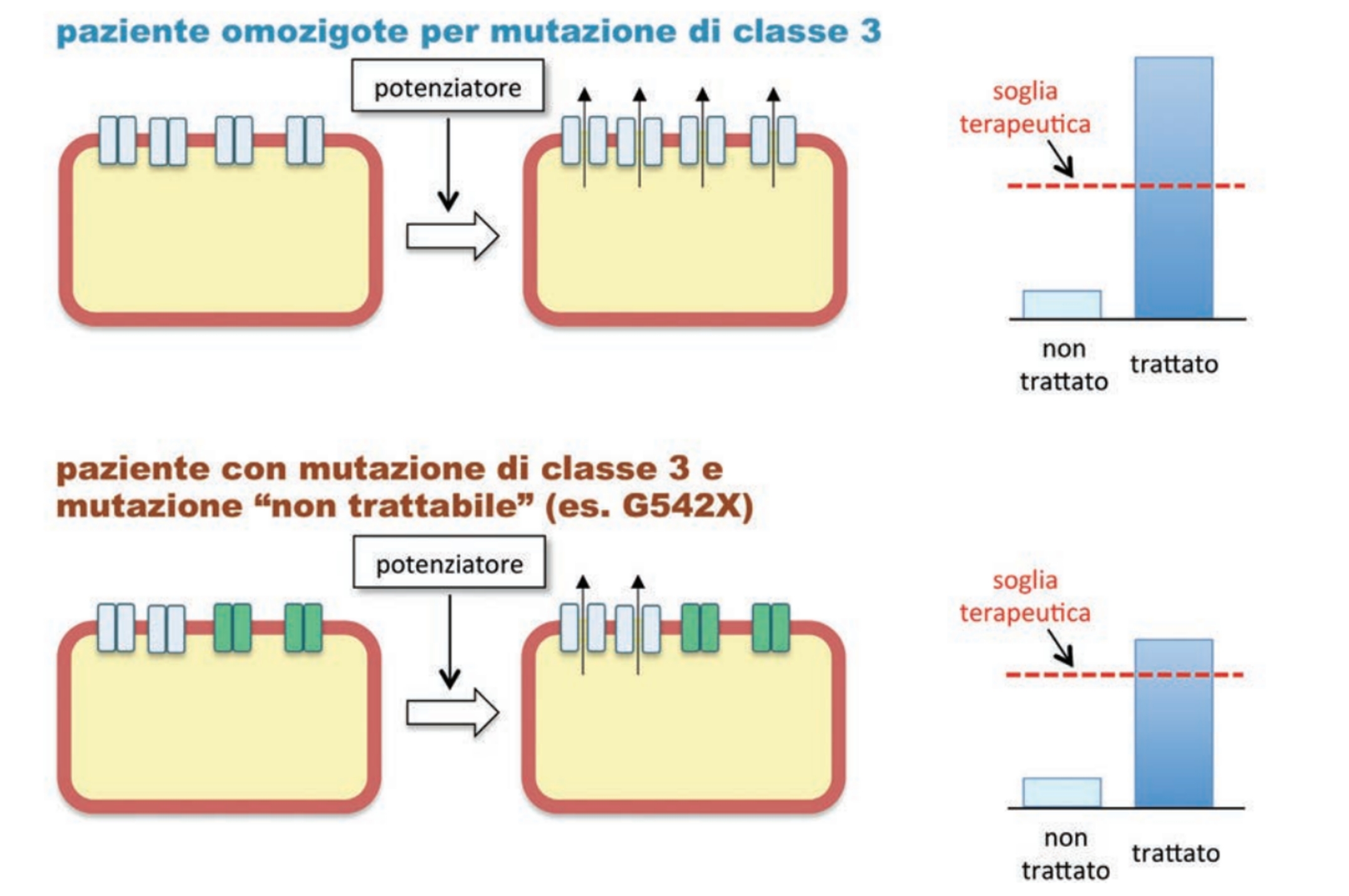
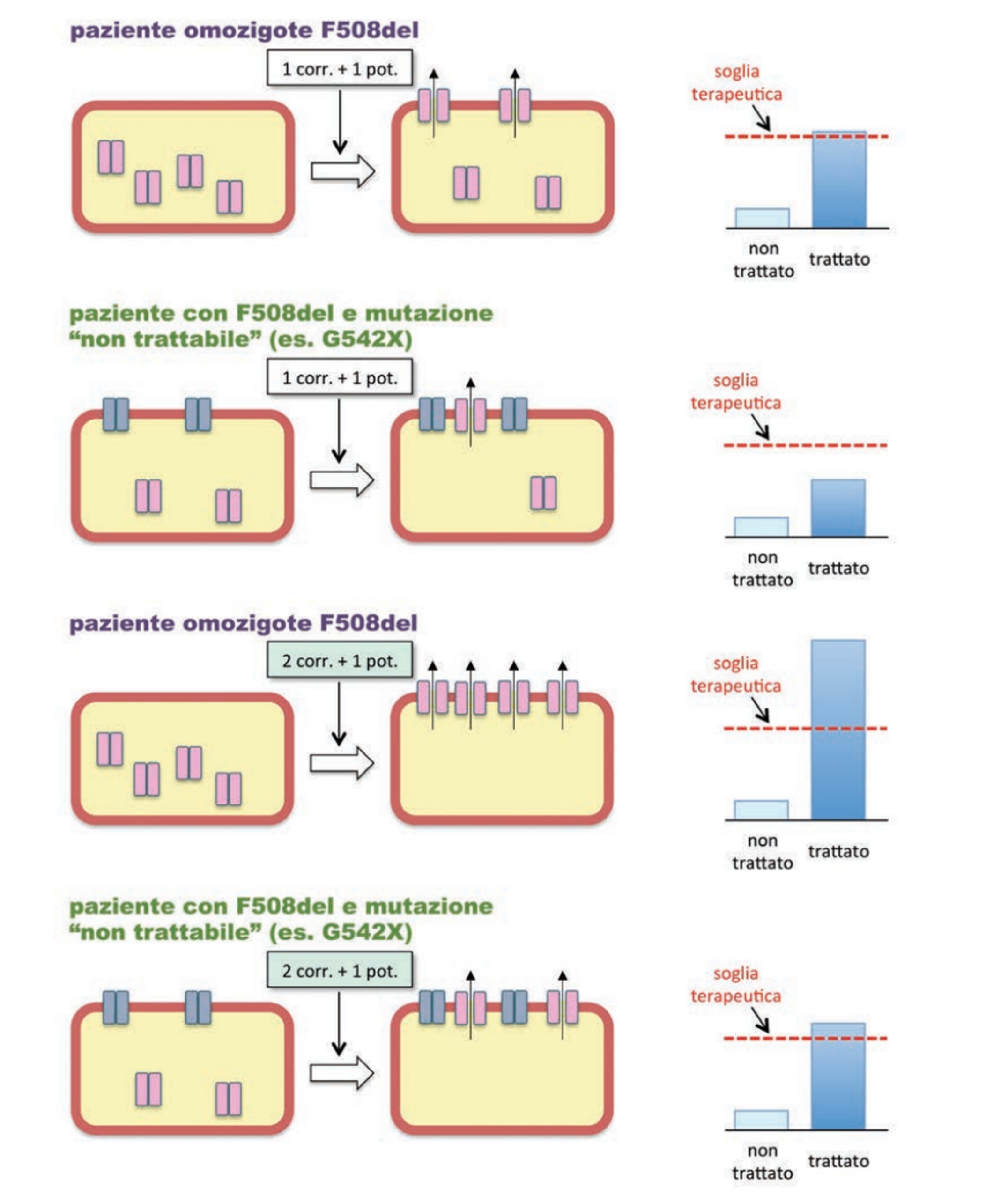
I modulatori della CFTR mutata sono distinti in base al meccanismo di azione e comprendono:
• potenziatori
• correttori
• amplificatori