Le mutazioni a carico del gene CFTR, responsabile della fibrosi cistica, determinano la formazione di secrezioni dense e viscose che danneggiano l’organismo a livello sistemico, ma colpiscono in particolar modo l’apparato respiratorio e digestivo. Benché al momento non sia possibile associare le singole mutazioni nel gene CFTR a sintomi specifici e neppure definire un iter prognostico certo per i pazienti affetti da fibrosi cistica, è chiaro che le manifestazioni cliniche variano con l’età.
I principali sintomi della fibrosi cistica sono: • tosse, espettorazione e dispnea. L’espettorazione può essere accompagnata da vomito • ileo da meconio, stipsi e occlusione intestinale • sudore salato, ricco di elettroliti • insufficienza pancreatica • malassorbimento e malnutrizione
Nella maggior parte dei casi la diagnosi di fibrosi cistica viene posta nelle prime settimane successive alla nascita, grazie allo screening neonatale o entro i primi due anni di vita.
Esistono tuttavia via casi in cui la diagnosi di fibrosi cistica viene posta in età adulta. Si tratta di casi in cui la manifestazione della malattia è molto lieve, grazie al mantenimento della funzionalità residua della CFTR. In questi casi i pazienti possono soffrire di infezioni respiratorie durante l’infanzia, ma solo successivamente, in età adulta sviluppano un quadro clinico più severo e tipico della fibrosi cistica: bronchiectasia, pancreatite o infertilità.
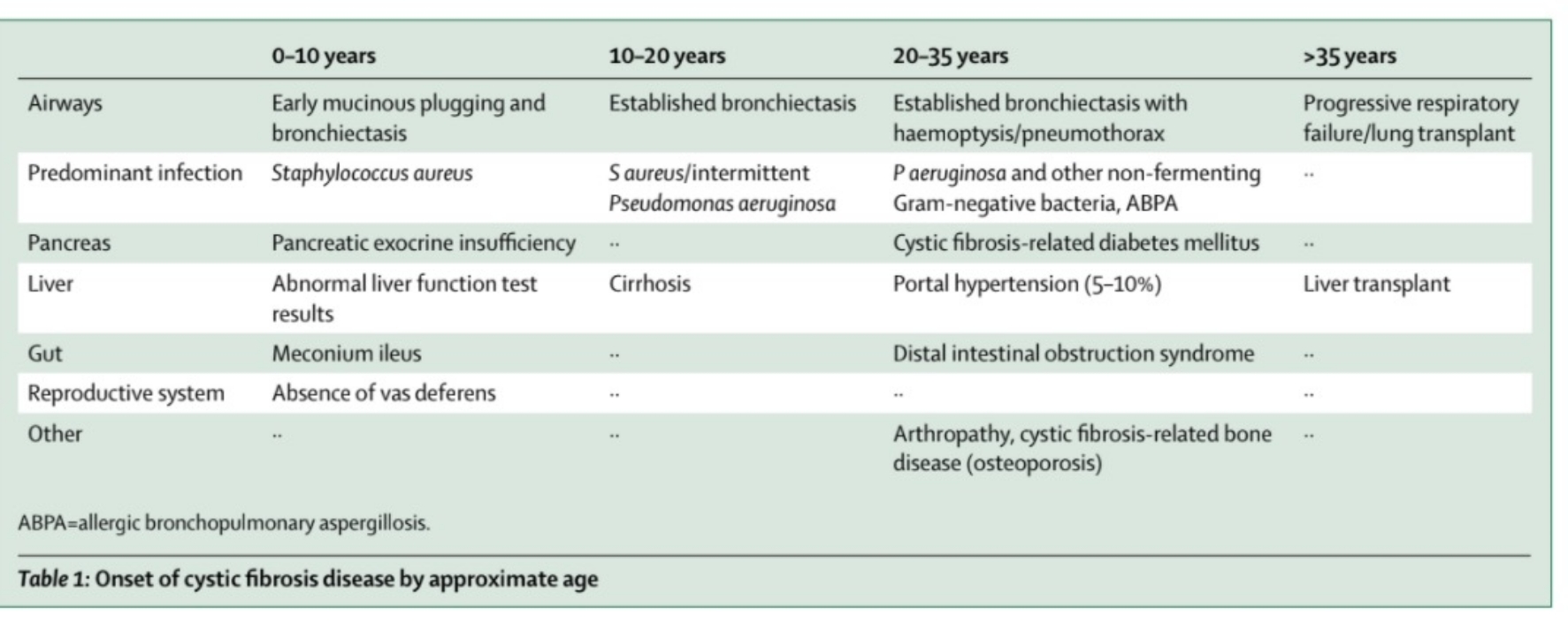
Tabella. Ipotesi tra manifestazione dei sintomi della fibrosi cistica ed età del paziente.
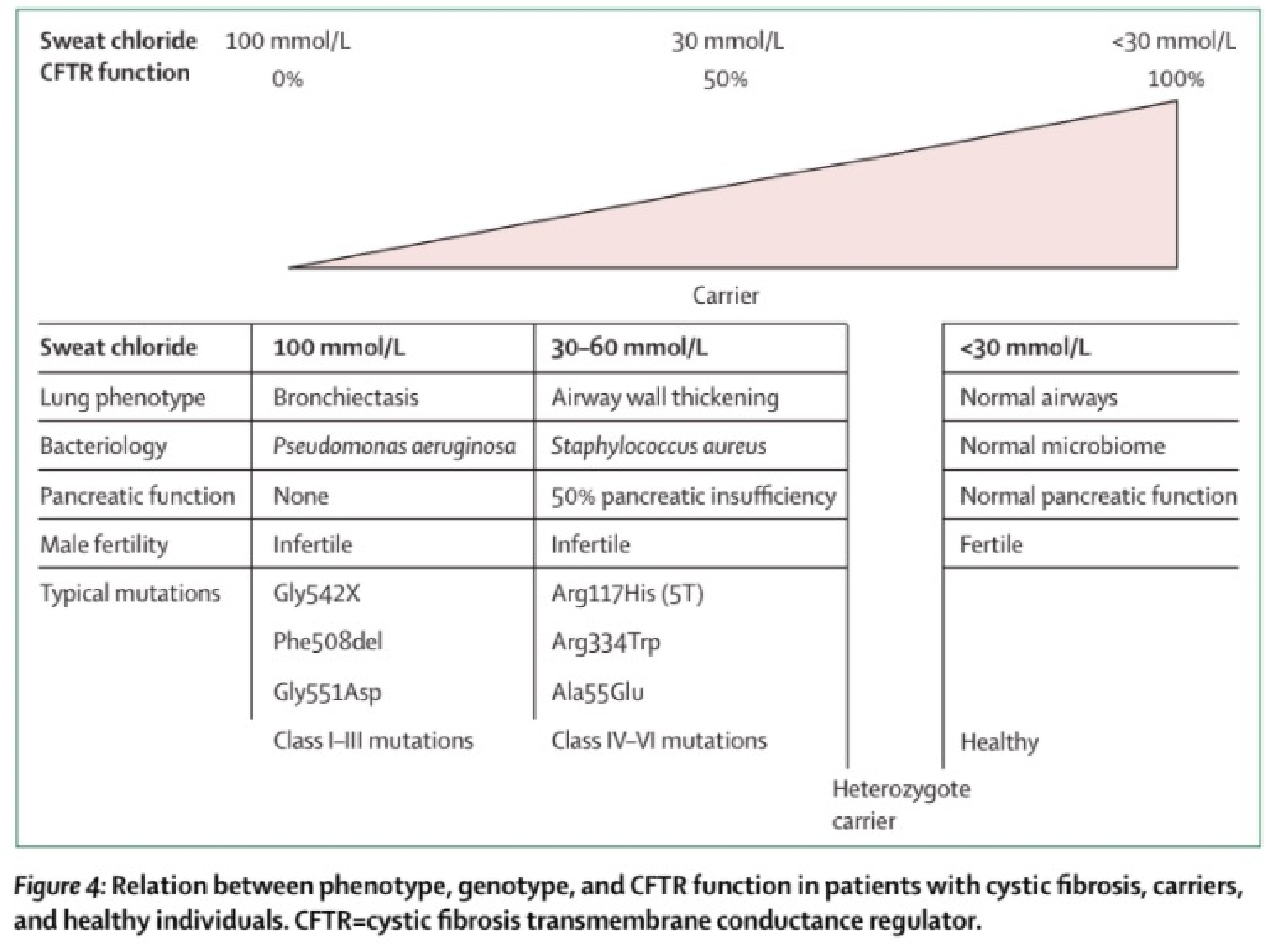
Figura. Correlazione tra genotipo, fenotipo e funzionalità della CFTR in pazienti con fibrosi cistica, portatori e soggetti sani.
Infiammazione e infezioni delle vie respiratorie
L’apparato respiratorio è il principale organo interessato dalla fibrosi cistica e tosse e catarro denso, anche purulento sono i principali sintomi.
Benché alla nascita i bambini possano avere polmoni istologicamente normali, per un terzo di essi la patologia polmonare esordisce con la comparsa di muco e infezioni respiratorie nelle prime settimane di vita. L’eccessiva produzione di muco, denso e viscoso ostruisce le vie aeree che diventano sede di infezioni batteriche ricorrenti e infiammazione cronica che progressivamente nel tempo portano a una perdita della funzionalità respiratoria. Il danno polmonare inizia probabilmente con una ostruzione diffusa a carico delle vie aeree di piccolo calibro a causa del muco eccessivamente denso e viscoso. L’esordio dei sintomi respiratori non è uguale in tutti i pazienti. I primi sintomi respiratori possono manifestarsi nelle prime settimane di vita o anche più tardi, durante l’adolescenza o in età adulta.
Alcuni segni possono indurre il sospetto di problema respiratorio, oltre alle infezioni batteriche: • tosse cronica, spesso a carattere pertussoide • aumento della frequenza del respiro • calo di appetito • rallentamento della crescita associato a diarrea cronica • perdita di peso • scarsa resistenza allo sforzo • malessere generale
I patogeni più frequenti
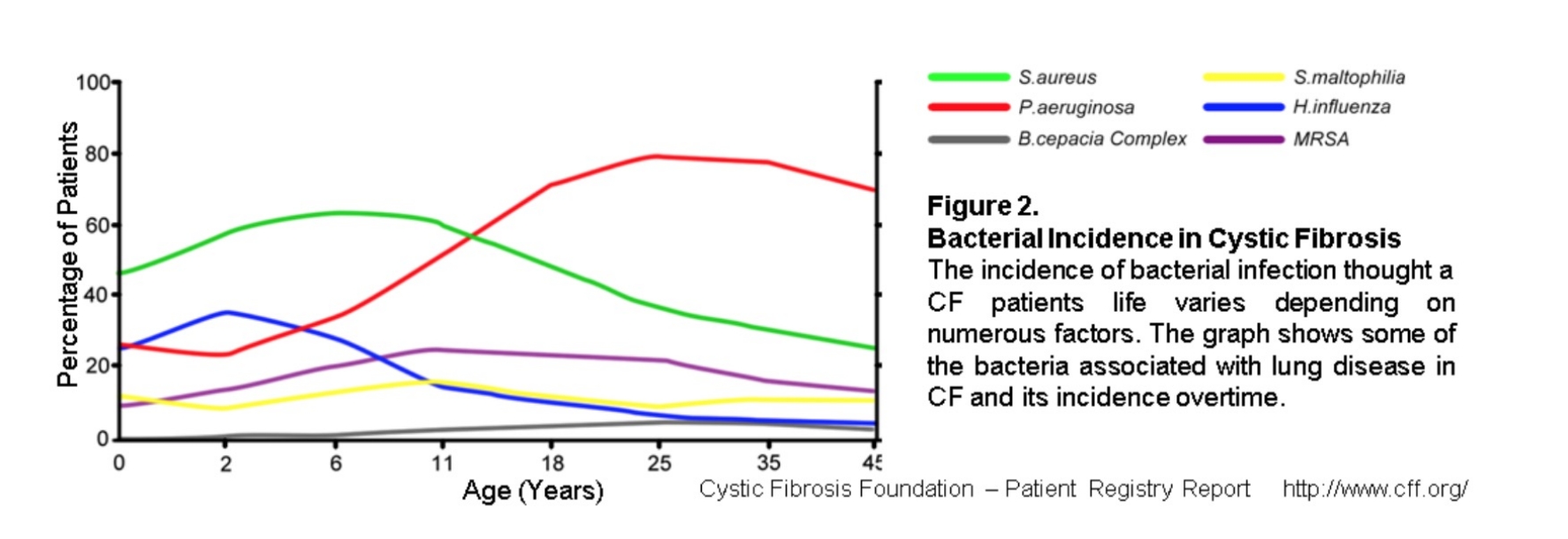
Inizialmente, soprattutto nell’infanzia, i patogeni che più frequentemente colonizzano il tratto respiratorio dei pazienti con fibrosi cistica sono Haemophilus influenzae e lo Staphylococcus aureus. Con il progredire della malattia, Pseudomonas aeruginosa è il batterio Gram-negativo più frequentemente isolato nei polmoni dei pazienti. Questo batterio opportunista è tipico delle infezioni nosocomiali e colpisce generalmente persone con difese immunitarie o integrità degli epiteli compromessi. Un ceppo mucoide di P. aeruginosa è associato unicamente alla fibrosi cistica e si associa a una peggior prognosi rispetto ai ceppi non mucoidi.Un altro patogeno degno di nota nella fibrosi cistica, per il maggior rischio di morte, è Burkholderia cepacia complex, riscontrato approssimativamente nel 2,6% dei pazienti. Oltre ai batteri Gram-negativi, sono potenziali patogeni respiratori per i pazienti con fibrosi cistica anche i micobatteri non tubercolari, in particolar modo Mycobacterium abscessus e Mycobacterium avium-intracellulare. La loro prevalenza nei pazienti con fibrosi cistica è in aumento e possono essere difficili da trattare per la loro resistenza costituzionale agli antibiotici. Infine, vale la pena nominare anche i batteri anaerobi obbligati. Essi sono presenti comunemente anche nelle vie aeree dei soggetti sani e per questo non è chiaro il loro ruolo nella patogenesi dei pazienti con fibrosi cistica.
Figura. I principali patogeni respiratori nei pazienti con fibrosi cistica.
Ileo da meconio e occlusione intestinale
L’ileo da meconio alla nascita e le ostruzioni intestinali nelle età successive sono i principali sintomi intestinali associati alla fibrosi cistica.
In entrambi i casi la causa è riconducibile all’insufficienza pancreatica esocrina con deficit di enzimi digestivi, con conseguente ristagno di materiale fecale (o del meconio) misto a muco.
L’ileo da meconio è presente approssimativamente nel 15% dei neonati affetti da fibrosi cistica, soprattutto negli omozigoti per la mutazione DF508. Il 90% dei neonati con ileo da meconio è affetto da fibrosi cistica. In alcuni casi il problema intestinale è già presente durante la gestazione e si può verificare perforazione intestinale e peritornite meconiale.
Si manifesta tipicamente con distensione addominale, vomito e incapacità di evacuare il meconio.
Nei bambini più grandi e anche negli adulti si possono verificare stipsi intermittente o cronica accompagnata da dolori addominali e occlusione intestinale (prevalenza 20-25%).
Altri problemi intestinali comprendono invaginazione, volvolo, prolasso rettale, ascesso periappendicolare.
Secondo studi recenti la fibrosi cistica sembra favorire un quadro infiammatorio intestinale e disbiotico tale da favorire l’insorgenza di malattie croniche intestinali come il morbo di Crohn e di neoplasie del tubo digerente, in particolare di carcinoma del colon.
Sudore salato
Il sudore ricco di elettroliti, cosiddetto sudore salato è una condizione presente nella quasi totalità dei pazienti (98-99%).
In seguito alla perdita o alla riduzione della funzionalità della proteina transmembrana CFTR (Cystic Fibrosis Tranmembrane Regulator) il sudore secreto risulta ricco di cloro (concentrazione patologica > 60 mmol/L), potassio e sodio.
La continua perdita di elettroliti con il sudore può comportare disidratazione iponatremica, ipokaliemia e alcalosi metabolica. Quest’ultima può rivelarsi particolarmente pericolosa nel neonato e nel lattante.
La perdita di sali, se non opportunamente compensata, può associarsi a inappetenza, vomito, torpore e agitazione e anche arresto della crescita.
Insufficienza pancreatica
La funzione pancreatica è compromessa nell’85% dei pazienti con fibrosi cistica. Nel 50% dei casi, l’insufficienza pancreatica si manifesta nei neonati già alla nascita o addirittura in utero e si associa a un genotipo caratterizzato dalle mutazioni più severe nel gene CFTR. Nei pazienti in cui la funzione della proteina CFTR è almeno parzialmente conservata l’insufficienza pancreatica può manifestarsi più tardivamente, generalmente nei primi anni di vita. In ogni caso, l’insufficienza pancreatica è il risultato del ristagno di secrezioni dense e ostruzione dei dotti pancreatici con conseguente collasso del tessuto e impossibilità di riversare gli enzimi nell’intestino.
In seguito all’assenza o riduzione degli enzimi pancreatici chimotripsina ed elastasi-1 grassi e proteine non vengono digerite (maldigestione). La carenza degli enzimi pancreatici si rende evidente con la presenza di feci abbondanti e untuose o saponose (steatorrea). Se non opportunamente trattata, alla maldigestione consegue malassorbimento dei nutrienti e malnutrizione con rallentamento della crescita in peso e altezza e ipovitaminosi. Già alla nascita i neonati con fibrosi cistica manifestano i segni di malassorbimento con un peso corporeo inferiore alla media e fatica nel recupero del calo ponderale.
Questa è la ragione principale per cui i bambini affetti da questa patologia hanno un accrescimento ritardato e scarso rispetto ai coetanei. Mantenere un buono strato di nutrizione è importante anche per la funzionalità respiratoria, è infatti nota la correlazione tra indice di massa corporea e il FEV1%.
E’ importante sottolineare che a causa dell’effetto compensatorio prodotto dalle amilasi e lipasi presenti nel latte materno, la sindrome da malassorbimento in alcuni neonati allattati al seno può essere molto lieve e difficilmente riconoscibile.
Nel 10-15% dei pazienti con insufficienza pancreatica lieve o pancreas funzionante, con genotipo caratterizzato da mutazioni meno severe (classi IV, V, VI) si possono verificare pancreatiti ricorrenti, che si manifestano con crisi ripetute e protratte di dolore addominale e innalzamento dei livelli di enzimi pancreatici nel sangue (amilasi pancreatica, tripsina e lipasi).
Bibliografia
• Radlovic N. Cystic fibrosis. Srp Arch Celok Lek 2012; 140(3-4):244-9
• Endres TM. What Is Cystic Fibrosis? JAMA. 2022 ;327(2):191
• De Boeck K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020;109(5):893-899
• Registro Italiano Fibrosi Cistica- Rapporto 2017-2018
• Elborn JS. Cystic fibrosis. Lancet 2016; 388(10059):2519-2531