La fibrosi cistica è una malattia genetica causata da un difetto del gene che codifica per la proteina di membrana CFTR (Cystic Fibrosis Transmembrane Regulator), la cui principale funzione è il trasporto transmembrana del cloro e di altri ioni. Il gene, identificato nel 1984 e clonato nel 1989, è localizzato sul braccio lungo del cromosoma 7 (7q31.2), è costituito da 27 esoni e codifica una proteina di 1480 aminoacidi che regola il trasporto di Cl– e HCO3– e indirettamente di Na+ negli epiteli e di conseguenza di acqua.
La proteina CFTR
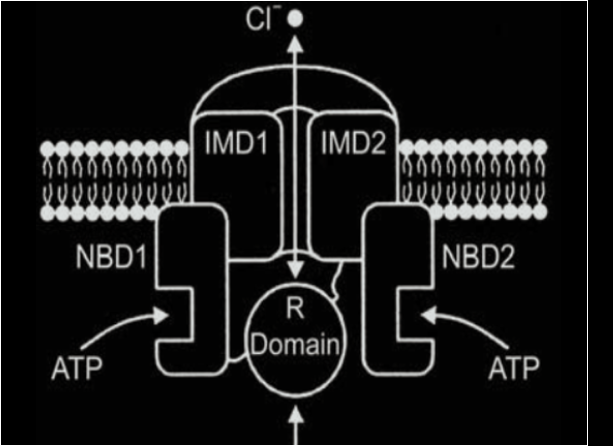
Il gene CFTR codifica per un canale transmembrana costituito da due domini transmembrana ognuno costituito da 6 alfa eliche (MSD, Membrane Spanning Domain). Ogni dominio MSD è connesso a un dominio citoplasmatico legante nucleotidi come ATP, chiamato Nucleotide Binding Domain (NBD). I nucleotidi sono importanti perché il canale è regolato da cAMP. Il dominio regolatore R (R-Domain) che regola l’apertura e chiusura del canale grazie a siti di fosforilazione delle protein-chinasi A e C, cAMP-dipendenti.
L’attivazione del canale CFTR è mediata da un agonista che induce un aumento di cAMP seguito dall’attivazione di una protein-chinasi A, che fosforila il dominio R. ll canale si apre ogni volta che un molecola di ATP si lega ai domini NBD1 e NBD2, mentre si chiude in seguito all’idrolisi di almeno uno dei due nucleotidi. Normalmente il CFTR è un canale che permette il passaggio del cloro all’esterno della cellula delle ghiandole esocrine dell’epitelio di rivestimento del tratto respiratorio, gastrointestinale e riproduttivo e regola negativamente il canale ENaC (canale epiteliale del sodio). In condizioni normali nella cellula entra poco sodio e pertanto viene mantenuta l’omeostasi.
Rappresentazione schematica del canale CFTR di trasporto transmembrana di ioni cloro e altri anioni.
Figura 1, tratta da Radlovic N. Cystic fibrosis. Srp Arch Celok Lek 2012; 140(3-4):244-9
Conseguenze dell’assenza o malfunzionamento del CFTR
Il canale CFTR si colloca sulla membrana cellulare di alcune ghiandole (salivari, pancreatiche e sudoripare), dei vasi deferenti, dell’intestino, delle vie respiratorie e delle vie biliari. Ad esclusione delle ghiandole sudoripare, se il canale è assente o mal funzionante, le cellule epiteliali secernono meno ioni Cl- e riassorbono Na+ in eccesso, determinando di conseguenza una riduzione di acqua. La conseguenza del difetto del canale CFTR è la produzione di secrezioni più asciutte, disidratate, dense e viscose. Nelle vie aeree, il muco più viscoso tende ad accumularsi lungo la parete bronchiale e la clearance mucociliare diventa inefficace, favorendo l’instaurarsi di infezione broncopolmonare e la tendenza al ripetersi di infezioni polmonari.
Nelle ghiandole sudoripare, il difetto del canale CFTR comporta invece un’incapacità della ghiandola a recuperare attraverso il suo dotto escretore il cloro e il sodio, che vengono prodotti all’origine in concentrazione uguale a quella del plasma. Ne consegue una secrezione di sudore ad alto contenuto di elettroliti e notoriamente “salato”.
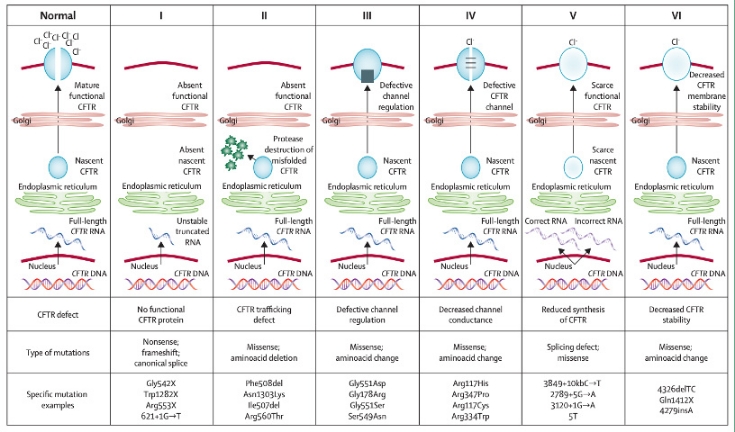
Il difetto della proteina CFTR non è uguale in tutti i malati affetti da fibrosi cistica. Può variare dalla totale mancanza della proteina stessa (difetto più grave), alla incompleta maturazione del canale o al funzionamento ridotto. I sintomi della malattia dipendono dal tipo di difetto e quindi dalla funzionalità del canale CFTR.
Figura 2, Elborn JS. Cystic fibrosis. Lancet 2016; 388(10059):2519-2531 ENaC: canali del sodio CFTR: Cystic Fybrosi Transmemabrane Regulator
CFTR: le mutazioni
Ad oggi sono state identificate oltre 2000 mutazioni differenti a livello del gene CFTR. La mutazione più frequente in tutte le popolazioni ed etnie è la F508del (DF508); in Italia la F508del rappresenta il 51% delle mutazioni. Le mutazioni del gene CFTR interessano solitamente una o poche basi nucleotidiche e sono distribuite principalmente negli esoni o nelle sequenze di giunzione tra esoni ed introni. Più raramente le mutazioni si trovano negli introni. La maggior parte delle mutazioni sono alterazioni missense, ma sono possibili e presenti anche frameshifts, difetti di splicing, mutazioni non-senso, inserzioni e anche delezioni in frame. Approssimativamente il 15% delle varianti genetiche identificate non è patologica. Mutazioni differenti possono alterare in diverso modo la funzionalità del canale del cloro CFTR e per questo determinare diverse forme di malattia. Al momento non è noto il tipo di difetto che ogni mutazione provoca nella proteina CFTR e pertanto non per tutte è nota la corrispondenza del quadro clinico e sintomatologico della fibrosi cistica.
Le mutazioni nel gene CFTR possono essere raggruppate in 6 classi, in base ai meccanismi biomolecolari responsabili del deficit funzionale della proteina: ● classe I: mutazioni che determinano l’assenza della proteina CFTR funzionante ● classe II: mutazioni che alterano il processamento della proteina. ● classe III: mutazioni che provocano un deficit nell’attivazione del canale, nella sua funzione di “gating”. Le mutazioni colpiscono i siti di legame dell’ATP ● classe IV: mutazioni che determinano un difetto della permeabilità ionica. Le mutazioni coinvolgono i domini transmembrana e provocano una diminuzione del flusso ionico e una riduzione del tempo di apertura del canale ● classe V: mutazioni nei siti di “splicing”, che riducono la quantità della proteina CFTR, ma non hanno un impatto sulla funzionalità. ● classe VI: la proteina CFTR è normalmente espressa ed attiva, ma va incontro a degradazione precoce.
Classificazione delle mutazioni nel gene CFTR.
Figura 3: tratta da Elborn JS. Cystic fibrosis. Lancet 2016; 388(10059):2519-2531
Le mutazioni CFTR, da genotipo a fenotipo
In generale i pazienti con combinazioni nei due alleli delle varianti patogenetiche di classe I, II e III, che determinano la perdita della funzionalità della proteina CFTR, manifestano per lo più un fenotipo severo della malattia, con caratteristiche cliniche che comprendono: ● insufficienza pancreatica, ● ileo da meconio alla nascita, ● malassorbimento e difetto di crescita, ● broncopneumopatia e compromissione della funzionalità respiratoria con andamento peggiorativo nel tempo, ● malattia epatica.
Le mutazioni appartenenti alle classi IV, V e VI sono in genere associate a un quadro clinico di fibrosi cistica più lieve con sufficienza pancreatica, compromissione polmonare più modesta e migliori aspettative di vita.
Le varianti patogenetiche di classe IV, V e VI sono fenotipicamente dominanti quando associate a varianti patogenetiche di classe I, II e III. Tuttavia, sia la variabilità genetica sia quella ambientale, nonché le terapie, rendono la correlazione genotipo – fenotipo complicata e spesso difficilmente prevedibile. Pertanto, la previsione del decorso della malattia a partire dal genotipo non è una prassi raccomandata. Spesso, infatti, pazienti con il medesimo genotipo possono presentare manifestazioni cliniche differenti, soprattutto per quanto concerne la compromissione polmonare. La presenza di varianti in cis del gene CFTR o di varianti in altri geni definiti come “geni modificatori”, di cui ancora non è definito il ruolo funzionale, potrebbe spiegare parte di queste discrepanze. Tali variazioni di sequenza sono state infatti associate, in alcuni casi, ad una più frequente insorgenza di specifiche manifestazioni cliniche. E’ importante sottolineare che non esiste al momento una chiara correlazione genotipo-fenotipo, poiché nella manifestazione della fibrosi cistica concorrono anche i cosiddetti geni modificatori e i fattori epigenetici. Non è quindi possibile prevedere un iter prognostico della malattia in base alle mutazioni di un paziente affetto da fibrosi cistica.
Bibliografia
• Radlovic N. Cystic fibrosis. Srp Arch Celok Lek 2012; 140(3-4):244-9 • De Boeck K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020;109(5):893-899 • Elborn JS. Cystic fibrosis. Lancet 2016; 388(10059):2519-2531 • Registro Italiano Fibrosi Cistica- Rapporto 2017-2018 • Analisi Genetica in FC – Consensus 2019 • Boucher RC. Annu Rev Med. 2007 • NHS. Cystic fibrosis.